What we care about the most…
Proteasomes
Protein homeostasis is essential for cellular survival. The main non-lysosomal protein degradation machinery in eukaryotic cells is the proteasome, a large macromolecule equipped with proteases. The core proteasome particle has been widely accepted to function intracellularly (cytosol and nucleus) and produce 3-30 amino acid-long peptide molecules as a result of protein breakdown. Beyond its conventional role in preserving protein homeostasis, the proteasome has been shown to be integral to neuronal functions such as synaptic transmission and plasticity.
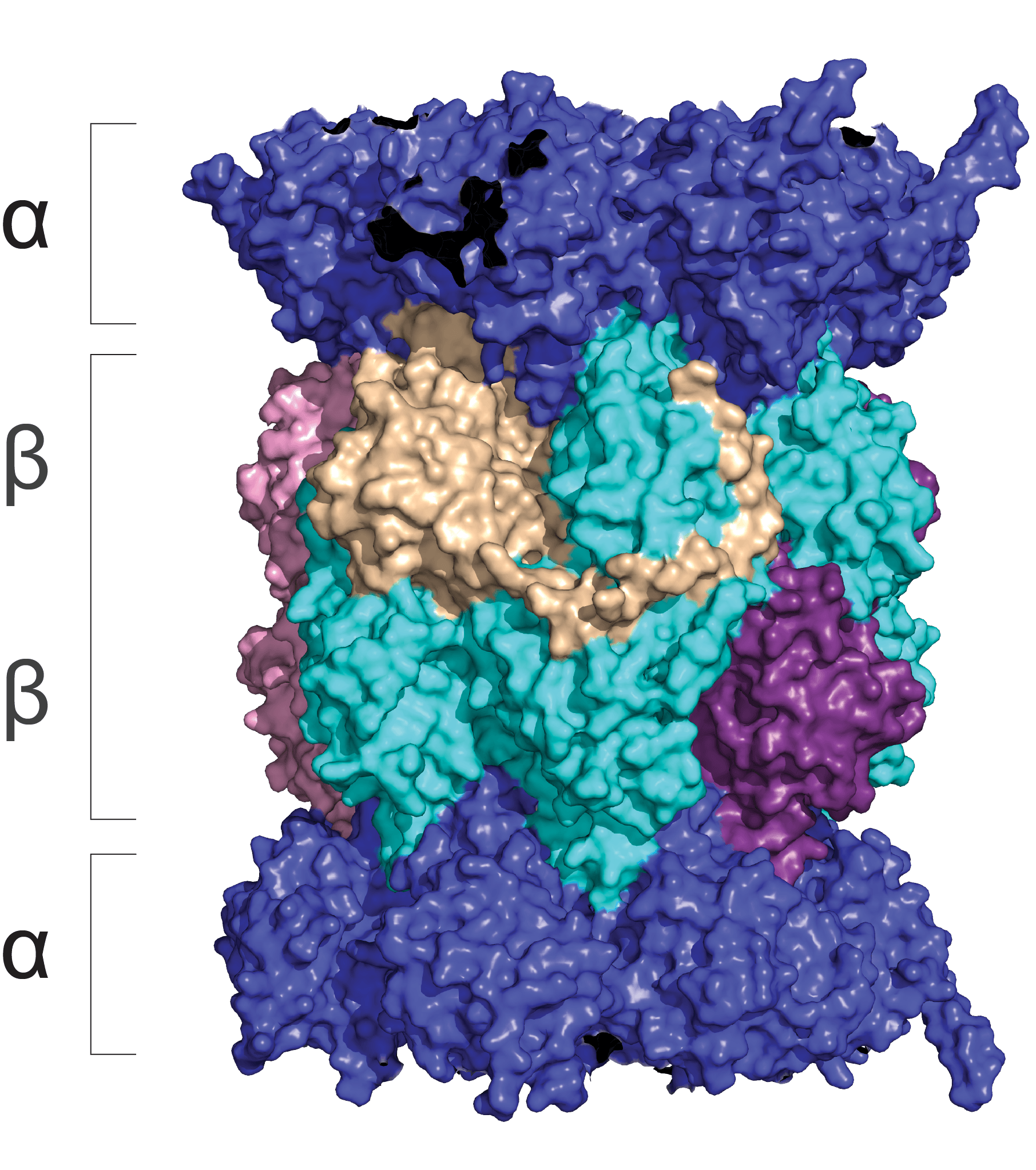
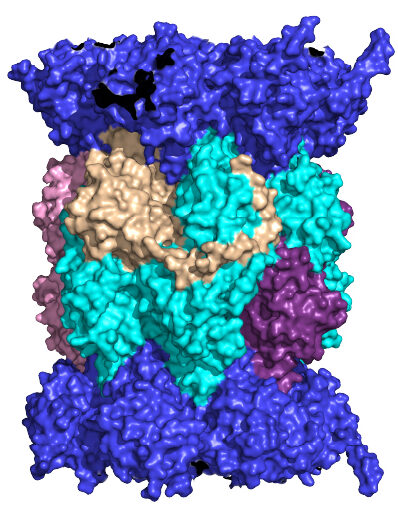
Neuronal membrane proteasomes (NMPs)
The proteasome has recently been shown to localize to the neuronal plasma membrane, forming a distinct pool referred to as the neuronal membrane proteasome (NMP). These membrane-tethered complexes are catalytically active upon neuronal stimulation and are uniquely positioned to release peptide products directly into the extracellular space. Our previous findings, along with emerging evidence, indicate that NMPs degrade newly synthesized intracellular proteins and generate bioactive extracellular peptides, suggesting a novel, co-translational degradation mechanism . These peptides are not merely byproducts; when applied to naïve neurons, they are sufficient to modulate neuronal signaling, pointing to a previously unrecognized mode of neuron-to-neuron communication. Notably, the surface localization of proteasomes is significantly reduced in models of Alzheimer’s disease and Huntington’s disease, raising the possibility that NMP dysfunction contributes to disease progression. Based on these observations, we propose that NMP and its peptide products act as endogenous modulators of neuronal signaling under physiological conditions, and that disruption of this pathway contributes to altered signaling dynamics in disease.
Huntington’s disease (HD)
Huntington’s disease (HD) is a rare but fatal neurodegenerative disorder caused by a CAG trinucleotide repeat expansion in the huntingtin (HTT) gene, leading to the production of mutant huntingtin protein (mHTT) with an abnormally extended polyglutamine (polyQ) tract. When the number of repeats exceeds 40, mHTT becomes prone to misfolding and aggregation, forming intracellular inclusion bodies over time. These aggregates contribute to progressive neurodegeneration, with prominent atrophy in the striatum and cerebral cortex. HD affects approximately 5–10 individuals per 100,000 in Western populations. Although the genetic cause of HD is well defined, the molecular mechanisms driving disease onset and progression are poorly understood. Similar to other neurodegenerative diseases such as Alzheimer’s and Parkinson’s, HD is characterized by disrupted proteostasis, contributing to neuronal vulnerability. Our goal is to investigate the role of the neuronal membrane proteasome (NMP)—a neuron-specific, plasma membrane-localized protein degradation machinery—in the context of HD pathogenesis and its potential involvement in early disease-associated signaling dysfunction.
Projects
Elucidating the regulation of the neuronal membrane proteasome (NMP) in the R6/2 mouse model of Huntington’s disease
Defining proteasome and NMP dynamics using compartmentalized microfluidic neuron culture platforms